BAUDOUIN, Roi des Belges,
A tous, présents et à venir, Salut.
Vu la loi du 26 mars 1971 sur la protection des eaux de surface contre la pollution
notamment l'article 3, § 2;
Vu l'accord européen sur la limitation de l'emploi de certains détergents dans les
produits de lavage et de nettoyage, fait à Strasbourg le 16 septembre 1968, approuvé par
la loi du 28 février 1970;
Vu la directive 73/404/CEE du 22 novembre 1973 du Conseil des Communautés européennes
concernant le rapprochement des législations des Etats membres relatifs aux détergents,
modifiée par les directives 82/242/CEE du 31 mars 1982 et 86/94/CEE du 10 mars 1986;
Vu la directive 73/405/CEE du 22 novembre 1973 du Conseil des Communautés européennes
concernant le rapprochement des législations des Etats membres relatives aux méthodes de
contrôle de la biodégradabilité des agents de surface anioniques, modifiée par la
directive 82/243/CEE du 31 mars 1982;
Vu la directive 82/242/CEE du 31 mars 1982 du Conseil des Communautés européennes
concernant le rapprochement des législations des Etats membres relatives aux méthodes de
contrôle de la biodégradabilité des agents de surface non ioniques et modifiant la
directive 73/404/CEE;
Vu l'avis du Conseil d'Etat;
Sur la proposition de Notre Premier Ministre et de Notre Secrétaire d'Etat à
l'Environnement et de l'avis de Nos Ministres qui en ont délibéré en Conseil,
Nous avons arrêté et arrêtons :
Article 1er. Pour l'application du présent arrêté, on entend par détergent, tout produit dont la composition a été spécialement étudiée pour concourir au développement des phénomènes de détergence et qui comprend des composants essentiels (agents de surface) et, généralement, des composants complémentaires (adjuvants, renforçateurs, charges, additifs et autres composants accessoires).
Art. 2 § 1er. Il est interdit d'importer, de mettre sur le marché et d'utiliser des détergents lorsque la biodégradabilité moyenne des agents de surface qui y sont contenus est inférieure à 90 % pour chacune des catégories cationiques et ampholytes, à 80 % pour chacune des catégories anioniques et non ioniques.
§ 2. Jusqu'au 31 décembre 1989, les produits suivants contenant des agents de surface non ioniques, peuvent ne pas être conformes aux conditions fixées au § ler :
1° les produits d'addition peu moussants d'oxydes d'alkènes sur des substances telles qu'alcools, alkylphénols, glycols, polyols, acides gras, amides et amines utilisés dans les produits pour lave-vaisselle;
2° les éthers d'alkyles et d'alkylarylpolyglycols bloqués en fin de chaîne et alcalinorésistants et substances des types visés sous 1° utilisés dans les produits de nettoyage destinés aux industries alimentaires et aux industries métallurgiques.
§ 3. La disposition du § 2 ne s'applique que si les agents visés dans ce paragraphe ont une biodégradabilité plus élevée que celle des produits existants destinés au même emploi.
§ 4. L'emploi des agents de surface non ioniques faisant l'objet de la dérogation temporaire en vertu du § 2 ne peut pas, dans les conditions normales d'utilisation, porter préjudice à la santé de l'homme ou de l'animal.
Art. 3. La conformité aux exigences de l'article 2 est constatée via les mesures de contrôle reprises en annexe Ier, pour les agents de surface anioniques et en annexe II pour les agents de surface non ioniques.
Art. 4. Les indications suivantes doivent figurer sur les emballages sous lesquels les détergents sont présentés au consommateur, en caractères lisibles, visibles et indélébiles :
a) dénomination du produit;
b) le nom ou la raison sociale et l'adresse, ou la marque déposée du responsable de l'importation ou de la mise sur le marché.
Ces mêmes indications doivent figurer sur les documents d'accompagnement des détergents transportés en vrac.
Art. 5. L'arrêté royal du 23 mars 1977, relatif aux taux de biodégradabilité de certains agents de surface dans les détergents, est abrogé.
Art. 6. Notre Premier Ministre et Notre Secrétaire d'Etat à l'Environnement sont chargés, chacun en ce qui le concerne, de l'exécution du présent arrêté.
Annexe I
DETERMINATION DE LA BIODEGRADABILITE
DES AGENTS DE SURFACE ANIONIQUES
Méthode de référence (test de confirmation)
CHAPITRE ler
1.1. Définition.
Aux termes du présent arrêté, les agents de surface anioniques sont les agents qui, après passage sur échangeurs d'ions cationiques et anioniques, sont séparés par élution fractionnelle et déterminés sous forme de substance active au bleu de méthylène (MBAS) suivant la méthode d'analyse décrite au chapitre 3.
1.2. Equipement nécessaire.
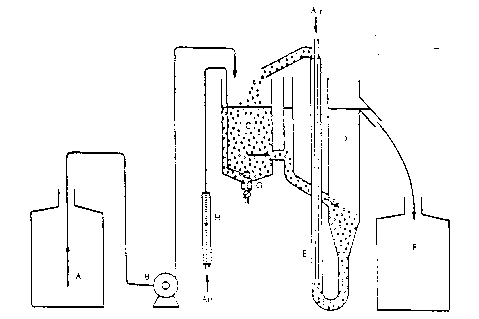
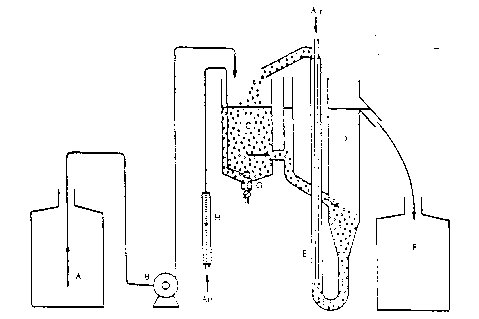
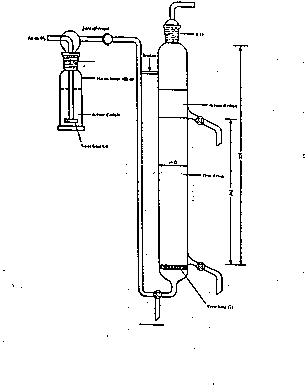
La méthode de mesure est fondée sur l'emploi d'une installation à boue activée, schématisée sur la figure 1 et représentée de manière plus détaillée sur la figure 2.
L'équipement se compose d'un récipient A, destiné à stocker les eaux résiduaires synthétiques, d'une pompe doseuse B, d'une cuve d'aération C, d'un décanteur D, d'une pompe à air comprimé E pour recycler la boue activée et d'un récipient F destiné à recueillir l'effluent traité.
Les récipients A et F doivent être en verre ou en matière plastique appropriée et contenir au moins 24 l. La pompe B doit assurer une alimentation régulière de la cuve d'aération en effluent synthétique, en cours de fonctionnement normal, cette cuve doit contenir 3 l de mélange. Un verre fritté G destiné à l'aération est suspendu dans la cuve C au sommet du cône intérieur de cette cuve. La quantité d'air insufflée par le dispositif d'aération doit être contrôlée par un débitmètre.
1.3. Effluent synthétique.
Pour effectuer cet essai, on se sert d'un effluent synthétique.
Dissoudre par litre d'eau du robinet :
160 mg de peptone;
110 mg d'extrait de viande;
30 mg d'urée [CO (NH2)2];
7 mg de chlorure de sodium (NaCl);
4 mg de chlorure de calcium (CaCl2 - 2H2O);
2 mg de sulfate de magnésium (MgSO4 - 7H2O);
28 mg de monohydrogénophosphate de potassium (K2HPO4);
20 ± 2 mg de MBAS.
On extrait la MBAS du produit faisant l'objet de l'essai par la méthode indiquée au chapitre 2. L'effluent synthétique est préparé chaque jour.
1.4. Préparation des échantillons.
1.4.1. Les agents de surface non formulés peuvent être essayés tels quels. La teneur en MBAS doit être déterminée afin de préparer l'effluent synthétique (1.3).
1.4.2. Dans le cas de formulations, on procède à la détermination de la teneur en MBAS et en savon. On procède à une extraction alcoolique et à une séparation de la MBAS (voir chapitre 2). Il faut connaître la teneur en MBAS de l'extrait pour préparer l'effluent synthétique.
1.5. Fonctionnement de l'installation.
Au départ, on remplit la cuve d'aération C et le décanteur D avec de l'effluent synthétique. Le décanteur D doit être fixé à une hauteur telle que la cuve d'aération C contienne 3 l.
On introduit 3 ml d'un effluent secondaire de bonne qualité, fraîchement prélevé dans une installation de traitements d'eaux résiduaires, essentiellement domestiques. L'effluent doit être maintenu dans des conditions aérobies pendant la période de prise entre l'échantillonnage et l'utilisation. On met ensuite en marche le dispositif d'aération, la pompe à air comprimé E et la pompe doseuse B. L'effluent synthétique doit passer dans la cuve d'aération C au débit horaire de 1 l, ce qui donne un temps moyen de rétention de trois heures.
Il faut régler le rythme d'aération de telle façon que le contenu de la cuve C reste constamment en suspension et que la teneur en oxygène dissous soit au minimum de 2 mg/l. La formation de mousse doit être empêchée par des moyens appropriés. On n'utilisera cependant pas d'agents antimousse qui ont une action inhibitrice sur la boue activée ou qui contiennent du MBAS. La pompe E doit être réglée de telle sorte qu'il y ait dans la cuve d'aération C un recyclage continu et régulier de la boue activée issue du décanteur. La boue qui s'est accumulée au sommet de la cuve d'aération C, au fond du décanteur D ou dans le circuit de circulation, doit être remise en circulation au moins une fois par jour par brossage ou par tout autre moyen approprié. Quand la boue ne décante pas, on peut favoriser la décantation par addition, répétée si nécessaire, de fractions de 2 ml d'une solution à 5 % de chlorure ferrique.
L'effluent sortant du décanteur D est recueilli dans la cuve F pendant vingt-quatre heures, après quoi on prélève un échantillon après avoir procédé à l'homogénéisation du mélange. La cuve F doit alors être nettoyée soigneusement.
1.6. Contrôle du dispositif de mesure.
La teneur en MBAS (en mg/l) de l'effluent synthétique est déterminée immédiatement avant usage.
La teneur en MBAS (en mg/l) de l'eau résiduaire collectée pendant vingt-quatre heures dans la cuve F doit être déterminée par la même méthode d'analyse, immédiatement après le prélèvement, sinon les échantillons sont conservés, de préférence par congélation. La concentration doit être déterminée à 0,1 mg MBAS/l près.
Pour vérifier la bonne marche de l'opération, on mesure au moins deux fois par semaine la demande chimique en oxygène (DCO) ou le carbone organique dissous (COD) de l'effluent filtré sur fibre de verre, accumulé dans la cuve F et de l'effluent synthétique filtré qui est stocké dans la cuve A.
La diminution de la DCO ou du COD doit se stabiliser lorsque la biodégradation journalière de la MBAS est à peu près régulière, c'est-à-dire à la fin de la période initiale indiquée sur la figure 3.
La teneur en matières sèches minérales de la boue activée contenue dans la cuve d'aération doit être déterminée deux fois par semaine (en g/l). Si elle dépasse 2,5 g/l, il faut éliminer l'excès de boue activée.
L'essai de biodégradation est effectué à la température ambiante; cette température doit être régulière et maintenue entre 292 et 297 K (19-24° C).
1.7. Calcul de la biodégradation.
Le pourcentage de biodégradation de la MBAS doit être calculé quotidiennement à partir de la teneur en MBAS (exprimée en mg/l) de l'effluent synthétique et de l'eau résiduaire correspondante recueillie dans la cuve F. Les valeurs ainsi obtenues doivent être représentées graphiquement, comme l'indique la figure 3.
La biodégradabilité de la MBAS est calculée en prenant la moyenne arithmétique des valeurs obtenues au cours des vingt et un jours suivant la période initiale, délai pendant lequel la biodégradation doit avoir été régulière et l'installation doit avoir fonctionné sans aucune perturbation. En aucun cas, la durée de la période initiale ne dépassera six semaines.
Les valeurs quotidiennes de la biodégradation doivent être calculées à 0,1 % près, mais le résultat final est déterminé au nombre entier près.
Dans certains cas, la fréquence des prélèvements peut être diminuée mais, pour calculer la moyenne, on utilisera les résultats d'au moins 14 prélèvements journaliers répartis sur la période de vingt et un jours qui suivent la période initiale.
CHAPITRE 2. - Traitement préliminaire des produits à examiner
2.1. Notes préliminaires.
2.1.1. Traitement des échantillons
Le traitement des agents de surface anioniques et des détergents préalablement à la détermination de la biodégradation par le test de confirmation est le suivant :
Produits |
Traitement |
| Agents de surface anioniques Détergents |
Néant Extraction alcoolique suivie d'une séparation par échange d'ions et d'une élution fractionnelle à partir de l'échangeur d'anions |
Le but de l'extraction alcoolique est d'éliminer des produits commercialisés les composants insolubles et inorganiques qui peuvent, le cas échéant, perturber le test de biodégradation.
2.1.2. Procédé d'échange d'ions.
Il est nécessaire, pour l'exactitude des tests de biodégradation, d'isoler et de séparer les agents de surface anioniques du savon et des agents de surface non ioniques et cationiques.
Ce résultat est obtenu grâce à l'application d'une technique d'échange d'ions utilisant une résine échangeuse d'anions macroporeuse et les agents d'élution appropriés permettant l'élution fractionnée. Le savon et les agents de surface anioniques et non ioniques se trouvent ainsi isolés en une seule opération.
2.1.3. Contrôle analytique.
Après homogénéisation, la teneur en agents de surface anioniques du détergent synthétique est déterminée suivant la méthode d'analyse à la MBAS. La teneur en savon est déterminée selon une méthode appropriée. Cette analyse des produits est nécessaire pour le calcul des quantités requises pour la préparation des fractions destinées aux essais de biodégradation.
Une extraction quantitative ne s'impose pas; toutefois, on extraira au moins 80 % des agents de surface anioniques. Habituellement, on en obtient 90 % et plus.
2.2. Principe.
A partir d'un échantillon homogène (poudres, pâtes et liquides desséchés), on obtient un extrait par l'éthanol qui contient les agents de surface, le savon et d'autres composants solubles de l'alcool, de l'échantillon de détergent.
L'extrait par l'éthanol est évaporé et dissout dans un mélange isopropanol/eau; on fait passer la solution ainsi obtenue à travers un dispositif mixte échange de cations fortement acide/échange d'anions macroporeux, porté à la température de 323 K (50° C). Cette température est nécessaire pour empêcher la précipitation des acides gras en milieu acide.
Les agents de surface non ioniques restent dans l'effluent.
Les acides gras du savon sont séparés par élution avec de l'éthanol contenant du dioxyde de carbone. On obtient alors les agents de surface anioniques sous forme de sels d'ammonium par élution avec une solution de bicarbonate d'ammonium dans un mélange isopropanol/eau. Ces sels d'ammonium sont utilisés pour le test de biodégradation.
Les agents de surface cationiques, susceptibles de perturber le test de biodégradation et la procédure analytique, sont éliminés par l'échangeur de cations placé au-dessus de l'échangeur d'anions.
2.3. Produits chimiques et appareillage.
2.3.1. Eau désionisée.
2.3.2. Ethanol, 95 % (v/v) C2H5OH (dénaturants admis méthyléthylcétone ou méthanol).
2.3.3. Mélange isopropanol/eau (50/50 v/v).
50 parties d'isopropanol (CH3CHOH-CH3)
50 parties d'eau (2.3.1.).
2.3.4. Solution de dioxyde de carbone dans l'éthanol (environ 0,1% de CO2); au moyen d'un tube de transfert doté d'un disque en verre fritté incorporé, faire barboter le dioxyde de carbone (CO2) à travers l'éthanol (2.3.2) pendant dix minutes. La solution doit être préparée extemporanément.
2.3.5. Solution de bicarbonate d'ammonium (60/40 v/v); 0,3 mol de NH4HCO3 dans 1000 ml d'un mélange isopropanol/eau constitué de 60 parties d'isopropanol et de 40 parties d'eau (2.3.1).
2.3.6. Echangeur de cation (KAT), fortement acide, résistant à l'alcool (50-100 mesh).
2.3.7. Echangeur d'anions (AAT), macroporeux, Merck Lewatit MP 7080 (70-150 mesh) ou équivalent.
2.3.8. Acide chlorhydrique, 10 % HCl (p/p).
2.3.9. Ballon à fond rond de 2 000 ml avec rodage conique et condenseur à reflux.
2.3.10. Entonnoir filtrant de 90 mm de diamètre (pouvant être chauffé) pour filtres en papier.
2.3.11. Fioles à vide de 2 000 ml.
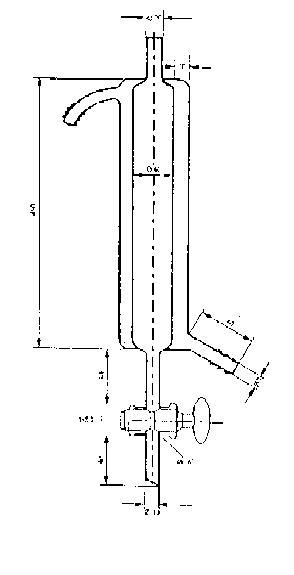
2.3.12. Colonnes d'échangeurs à enveloppe chauffante et robinet; tube intérieur de 60 mm de diamètre et de 450 mm de hauteur (figure 4).
2.3.13. Bain-marie.
2.3.14. Etuve de séchage à vide.
2.3.15. Thermostat.
2.3.16. Evaporateur rotatif.
2.4. Extraction et séparation des agents de surface anioniques.
2.4.1. Préparation de l'extrait.
La quantité d'agents de surface nécessaire pour l'essai de biodégradation est d'environ 50 g MBAS.
Normalement la quantité de produit à extraire ne dépasse pas 1000 g; cependant il peut être nécessaire d'extraire des quantités supplémentaires de l'échantillon.
Pour des raisons pratiques, la quantité de produit utilisé sera dans la plupart des cas limitée à 5 000 g lors de la préparation des extraits pour l'essai de biodégradation.
L'expérience a montré qu'une série d'extractions limitées est préférable à une seule extraction d'une grande quantité de produit. En ce qui concerne les échangeurs, les quantités spécifiées sont conçues pour une capacité de 600 à 700 mmol d'agents de surface et de savon.
2.4.2. Isolement des composants solubles dans l'alcool.
Ajouter 250 g du détergent à analyser à 1250 ml d'éthanol et porter le mélange à ébullition puis le soumettre à reflux pendant une heure, en agitant. Filtrer la solution alcoolique chaude dans un entonnoir filtrant à larges pores, porté à la température de 323 K (50° C) et aspirer fortement. Laver la fiole et l'entonnoir filtrant avec environ 200 ml d'éthanol chaud. Recueillir le filtrat et le lavage du filtre dans une fiole à vide.
Lorsque les produits à analyser sont des pâtes ou des liquides, s'assurer que l'échantillon ne contient pas plus de 55 g d'agents de surface anioniques et 35 g de savon. Evaporer cet échantillon pesé jusqu'à dessiccation complète. Dissoudre le résidu dans 2 000 ml d'éthanol et procéder comme ci-dessus.
Dans le cas de poudres de faible densité apparente ( < 300 g/l), il est recommandé d'augmenter la proportion d'éthanol dans le rapport 20/l.
Evaporer le filtrat d'éthanol jusqu'à dessiccation complète, de préférence au moyen d'un évaporateur rotatif. Répéter l'opération si une plus grande quantité d'extrait est nécessaire. Dissoudre le résidu dans 5 000 ml d'un mélange isopropanol/eau.
2.4.3. Préparation de colonnes d'échangeurs d'ions.
Colonne d'échangeurs de cations.
Placer 600 ml de résine échangeuse de cations (2.3.6) dans un bécher de 3 000 ml et couvrir en ajoutant 2 000 ml d'acide chlorhydrique (2.3.8). Laisser reposer pendant au moins deux heures en agitant de temps en temps. Décanter l'acide et transférer la résine dans la colonne (2.3.12) au moyen d'eau désionisée. La colonne doit comporter un tampon de laine de verre. Laver la colonne avec de l'eau désionisée à un débit de 10-30 ml/min jusqu'à ce que l'éluat soit exempt de chlorure. Déplacer l'eau avec 2 000 ml d'un mélange isopropanol/eau (2.3.3) à un débit de 10-30 ml/min. La colonne d échangeurs est prête à l'emploi.
Colonne d'échangeurs d'anions.
Placer 600 ml de résine échangeuse d'anions (2.3.7) dans un bécher de 3 000 ml et couvrir en ajoutant 2 000 ml d'eau désionisée. Laisser l'échangeur gonfler pendant au moins deux heures. Transférer la résine dans la colonne avec de l'eau. La colonne doit comporter un tampon en laine de verre désionisée comme couche de soutien des échangeurs.
Laver la colonne avec une solution 0,3 mol de bicarbonate d'ammonium (2.3.5) jusqu'à ce qu'elle soit exempte de chlorure, ce qui nécessite environ 5 000 ml de solution. Laver ensuite avec 2 000 ml d'eau désionisée. Déplacer l'eau avec 2 000 ml d'un mélange isopropanol/eau (2.3.3) à un débit de 10-30 ml/min. La colonne d'échangeurs est maintenant sous forme OH et prête à l'emploi.
2.4.4. Procédé d'échange d'ions.
Monter les colonnes d'échangeurs de sorte que la colonne d'échangeurs de cations se trouve au-dessus de la colonne d'échangeurs d'anions. Porter les colonnes à la température de 323 K (50° C) au moyen d'un thermostat. Chauffer 5 000 ml de la solution obtenue au point 2.4.2 jusqu'à 333 K (60° C) et passer la solution à travers le groupe d'échangeurs à un débit de 20 ml/min. Laver les colonnes avec 1 000 ml d'un mélange chaud d'isopropanol/eau (2.3.3).
Pour obtenir les agents de surface anioniques synthétiques (MBAS), démonter la colonne KAT. Eluer les acides gras du savon de la colonne KAT au moyen de 5 000 ml d'une solution d'éthanol/CO2 (à 323 K; 50° C) (2.3.4). Rejeter l'éluat.
Eluer ensuite les MBAS de la colonne AAT au moyen de 5 000 ml de solution de bicarbonate d'ammonium (2.3.5). Evaporer l'éluat jusqu'à dessication sur un bain de vapeur ou dans un évaporateur rotatif. Les résidus contiennent les MBAS (sous forme de sel d'ammonium) et, éventuellement, les produits anioniques non tensio-actifs qui ne nuisent pas au test de biodégradation. Ajouter de l'eau désionisée jusqu'à obtention d'un volume déterminé et doser la teneur en MBAS dans une fraction aliquote conformément au chapitre 3. La solution est utilisée comme solution étalon des détergents anioniques pour l'essai de biodégradation. La solution doit être maintenue à une température inférieure à 278 K (5° C).
2.4.5. Régénération des résines échangeuses d'ions.
L'échangeur de cations est jeté après l'emploi.
On régénère la résine échangeuse d'anions en faisant passer dans la colonne une quantité supplémentaire d'une solution de bicarbonate d'ammonium (2.3.5) à un débit d'environ 10 ml/min jusqu'à ce que l'éluat soit exempt d'agents de surface anioniques (essai au bleu de méthylène). Laver ensuite l'échangeur d'anions avec 2 000 ml d'un mélange isopropanol/eau (2.3.3). L'échangeur d'anions peut de nouveau être utilisé.
CHAPITRE 3. - Détermination des agents de surface anioniques dans l'essai de biodégradation
3.1. Principe.
La méthode est basée sur le fait que le colorant cationique qu'est le bleu de méthylène donne avec les agents de surface anioniques des sels bleus que l'on peut extraire au chloroforme. Afin d'éviter les interférences, l'extraction est d'abord effectuée à partir d'une solution alcaline et l'extrait est ensuite agité avec une solution acide de bleu de méthylène. L'absorbance de la phase organique séparée est mesurée par photométrie à la longueur d'onde d'absorption maximale de 650 nm.
3.2. Réactifs et appareillage.
3.2.1. Solution tampon ph 10 : dissoudre 24 g de bicarbonate de sodium (NaHCO3) pour analyse et 27 g de carbonate de sodium anhydre (Na2CO3) pour analyse dans de l'eau désionisée et diluer à 1000 ml.
3.2.2. Solution neutre de bleu de méthylène, dissoudre 0,35 g de bleu de méthylène pour analyse dans de l'eau désionisée et diluer 1 000 ml. Préparer la solution au moins vingt-quatre heures avant l'emploi.
L'absorbance de la phase chloroforme de l'essai à blanc, comparée à celle du chloroforme pur, ne doit pas dépasser 0,015 pour 1 cm d'épaisseur de la couche à 650 nm.
3.2.3. Solution acide de bleu de méthylène, dissoudre 0,35 g de bleu de méthylène pour analyse dans 500 ml d'eau désionisée et mélanger avec 6,5 ml d'H2SO4 (d = 1,84 g/ml). Diluer avec 1 000 ml d'eau désionisée. Préparer la solution au moins vingt-quatre heures avant l'emploi. L'absorbance de la phase de chloroforme de l'essai à blanc, comparée à celle du chloroforme pur, ne doit pas dépasser 0,015 pour 1 cm d'épaisseur de la couche à 650 nm.
3.2.4. Chloroforme (trichlorométhane) CHCl3 pour analyse, fraîchement distillé.
3.2.5. Dodécylbenzène-méthylester d'acide sulfonique.
3.2.6. Solution d'hydroxyde de potassium dans l'éthanol, KOH 0,1 mol.
3.2.7. Ethanol pur (C2H5OH).
3.2.8. Acide sulfurique (H2SO4 0,5 mol).
3.2.9. Solution de phénolphthaléine : dissoudre 1 g de phénolphtaléine dans 50 ml d'éthanol et ajouter 50 ml d'eau désionisée en agitant continuellement. Eliminer par filtration tout précipité obtenu.
3.2.10. Acide chlorhydrique et méthanol : 250 ml d'acide chlorhydrique concentré pour analyse et 750 ml de méthanol.
3.2.11. Ampoule à décantation de 250 ml.
3.2.12. Fiole jaugée de 50 ml.
3.2.13. Fiole jaugée de 500 ml.
3.2.14. Fiole jaugée de 1000 ml.
3.2.15. Ballon à fond rond avec rodage en verre, condenseur à reflux de 250 ml; granulés pour faciliter l'ébullition.
3.2.16. pH-mètre.
3.2.17. Photomètre pour mesures à 650 nm, avec des cuves de 1 à 5 cm.
3.2.18. Papier filtre qualitatif.
3.3. Méthode.
Les échantillons destinés à l'analyse ne doivent pas être prélevés à travers une couche de mousse.
Après avoir été soigneusement nettoyé à l'eau, l'appareillage utilisé pour l'analyse doit être entièrement rincé avec une solution d'acide chlorhydrique et de méthanol (3.2.10), puis avec de l'eau désionisée avant usage.
Filtrer les effluents d'entrée et de sortie de l'installation à boue activée à examiner dès l'échantillonnage. Eliminer les premiers 100 ml des filtrats.
Placer un volume mesuré de l'échantillon, neutralisé si nécessaire, dans une ampoule à décantation de 250 ml (3.2.11). Le volume de l'échantillon doit contenir entre 20 et 150 µg de MBAS. Pour une teneur en MBAS plus faible, on peut utiliser jusqu'à 100 ml de l'échantillon. Lorsqu'on en utilise moins de 100 ml, diluer à 100 ml avec de l'eau désionisée. Ajouter à l'échantillon 10 ml de la solution tampon (3.2.1) 5 ml de la solution neutre de bleu de méthylène (3.2.2) et 15 ml de chloroforme (3.2.4). Agiter le mélange de façon régulière et sans trop de vigueur pendant une minute. Après la séparation de phases, faire passer la couche de chloroforme dans une seconde ampoule à décantation contenant 110 ml d'eau désionisée et 5 ml de solution acide de bleu de méthylène (3.2.3). Agiter le mélange pendant une minute. Faire passer la couche de chloroforme sur un filtre de coton hydrophile préalablement lavé à l'alcool et imbibé de chloroforme dans une fiole graduée (3.2.12).
Extraire à trois reprises les solutions alcalines et acides, au moyen de 10 ml de chloroforme lors de la deuxième et de la troisième extraction. Filtrer les extraits combinés de chloroforme à travers le même filtre de coton hydrophile et diluer à la marque dans la fiole de 50 ml (3.2.12) avec le chloroforme utilisé pour relaver le coton hydrophile. Mesurer l'absorbance de la solution de chloroforme avec un photomètre à 650 nm dans des cuves de 1 à 5 cm en comparant avec celle du chloroforme pur. Faire un essai de dosage en blanc tout au long de la méthode.
3.4. Courbe d'étalonnage.
Préparer une solution d'étalonnage à partir de la substance étalon de dodécylbenzène-méthylester d'acide sulfonique (tétrapropylène type PM 340) après saponification dans le sel de potassium. La MBAS est exprimée en dodécylbenzène sulfonate de sodium (PM 348).
Peser 400 à 450 mg de dodécylbenzène-méthylester d'acide sulfonique (3.2.5) à 0,1 mg près dans un ballon à fond rond et ajouter 50 ml de solution d'hydroxyde de potassium et d'éthanol (3.2.6) et quelques granulés pour faciliter l'ébullition. Après avoir monté le condenseur à reflux, faire bouillir pendant une heure. Après refroidissement, laver le condenseur et le rodage de verre avec environ 30 ml d'éthanol et ajouter ces lavages au contenu du ballon. Titrer la solution à l'acide sulfurique jusqu'à décoloration de la phénolphtaléine. Transférer cette solution dans une fiole jaugée de l 000 ml (3.2.14), diluer à la marque avec de l'eau désionisée et mélanger.
Rediluer ensuite une partie de cette solution mère de l'agent de surface. En prélever 25 ml et transférer dans une fiole jaugée de 500 ml (3.2.13), diluer à la marque avec de l'eau désionisée et mélanger.
Cette solution étalon contient E x 1,023 mg MBAS/ml, E représentant le poids de l'échantillon en mg.
20 000
Pour établir la courbe d'étalonnage, prélever respectivement 1, 2, 4, 6 et 8 ml de la solution étalon et diluer chacun de ces prélèvements à 100 ml avec de l'eau désionisée. Procéder ensuite comme indiqué au point 3.3 (y compris un essai de dosage en blanc).
3.5. Calcul des résultats.
La courbe d'étalonnage (3.4) indique la quantité d'agent de surface anionique (MBAS) contenue dans l'échantillon. La teneur en MBAS de l'échantillon est indiquée par :
mg MBAS x 1000 = MBAS mg/l V
V = le volume en ml de l'échantillon utilisé.
Exprimer les résultats en dodécylbenzène sulfonate de sodium (PM 348).
3.6. Expression des résultats.
Exprimer les résultats en MBAS mg/l à 0,1 mg près.
Figure 1
| A Récipient de stockage | E Pompe à air comprimé |
| B Pompe doseuse | F Collecteur |
| C Cuve d'aération (capacité 3 l) | G Aérateur (verre fritté) |
| D Décanteur | H Débitmètre à air |
Figure 2
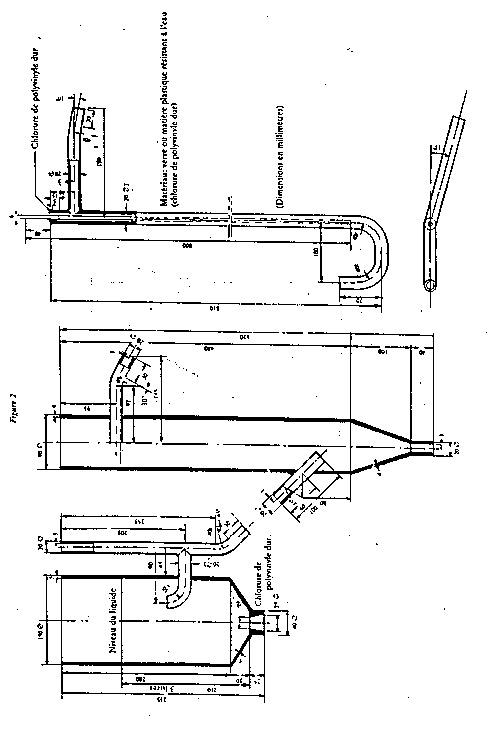
Figure 3
Calcul de la biodégradabilité - Test de confirmation
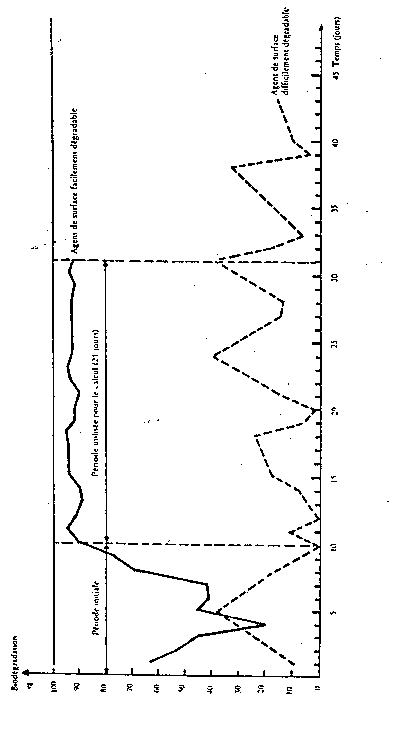
Figure 4
Calcul d'échangeur chauffant
(Dimensions en milimètres)
Vu pour être annexé à Notre arrêté du 25 octobre 1988.
Annexe II
DETERMINATION DE LA BIODEGRADABILITE
DES AGENTS DE SURFACE NON IONIQUES
Méthode de référence (test de confirmation)
CHAPITRE ler
1.1. Définition.
Aux termes du présent arrêté, les agents de surface non ioniques sont les agents qui, après passage sur échangeurs d'ions cationiques et anioniques, sont déterminés comme substance active au bismuth (BiAS) suivant la méthode d'analyse décrite au chapitre 3.
1.2. Equipement nécessaire.
La méthode de mesure est fondée sur l'emploi d'une installation à boue activée, schématisée sur la figure 1 et représentée de manière plus détaillée sur la figure 2.
L'équipement se compose d'un récipient A, destiné à stocker les eaux résiduaires synthétiques, d'une pompe doseuse B, d'une cuve d'aération C, d'un décanteur D, d'une pompe à air comprimé E pour recycler la boue activée et d'un récipient F destiné à recueillir l'effluent traité.
Les récipients A et F doivent être en verre ou en matière plastique appropriée et contenir au moins 24 l. La pompe B doit assurer une alimentation régulière de la cuve d'aération en effluent synthétique; en cours de fonctionnement normal, cette cuve doit contenir 3 l du mélange. Un verre fritté G destiné à l'aération est suspendu dans la cuve C au sommet du cône intérieur de cette cuve. La quantité d'air insufflée par le dispositif d'aération doit être contrôlée par un débitmètre H.
1.3. Effluent synthétique.
Pour effectuer cet essai, on se sert d'un effluent synthétique.
Dissoudre par litre d'eau du robinet :
160 mg de peptone;
110 mg d'extrait de viande;
30 mg d'urée [CO (NH2)2];
7 mg de chlorure de sodium (NaCl);
4 mg de chlorure de calcium (CaCl2 -2H2O);
2 mg de sulfate de magnésium (MgSO4 - 7H2O);
28 mg de monohydrogénophosphate de potassium (K2HPO4);
10 ± 1 mg de BiAS.
On extrait le BiAS du produit faisant l'objet de l'essai par la méthode indiquée au chapitre 2. L'effluent synthétique est préparé chaque jour.
1.4. Préparation des échantillons.
1.4.1. Les agents de surface non formulés peuvent être essayés tels quels. La teneur en BiAS doit être dosée afin de préparer l'effluent synthétique (1.3).
1.4.2. Dans le cas de formulations, on procède à la détermination des teneurs en BiAS, MSAS et en savon. On procède à une extraction alcoolique et à une séparation de la BiAS (voir chapitre 2). Il faut connaître la teneur en BiAS de l'extrait pour préparer l'effluent synthétique.
1.5. Fonctionnement de l'installation.
Au départ, on remplit la cuve d'aération C et le décanteur D avec de l'effluent synthétique. Le décanteur D doit être fixé à une hauteur telle que la cuve d'aération C contienne 3 l.
On introduit 3 ml d'un effluent secondaire de bonne qualité, fraîchement prélevé dans une installation de traitements d'eaux résiduaires, essentiellement domestiques. L'effluent doit être maintenu dans des conditions aérobies pendant la période de prise entre l'échantillonnage et l'utilisation. On met ensuite en marche le dispositif d'aération G, la pompe à air comprimé E et la pompe doseuse B. L'effluent synthétique doit passer dans la cuve d'aération C au débit horaire de 1 l, ce qui donne un temps moyen de rétention de trois heures.
Il faut régler le rythme d'aération de telle façon que le contenu de la cuve C reste constamment en suspension et que la teneur en oxygène dissous soit au minimum de 2 mg/l. La formation de mousse doit être empêchée par des moyens appropriés. On n'utilisera cependant pas d'agents antimousse qui ont une action inhibitrice sur la boue activée ou qui contiennent du BiAS. La pompe E doit être réglée de telle sorte qu'il y ait dans la cuve d'aération C un recyclage continu et régulier de la boue activée issue du décanteur. La boue qui s'est accumulée au sommet de la cuve d'aération C, au fond du décanteur D ou dans le circuit de circulation, doit être remise en circulation au moins une fois par jour par brossage ou tout autre moyen approprié. Quand la boue ne décante pas, on peut favoriser la décantation par addition, répétée si nécessaire, de portions de 2 ml d'une solution à 5 % de chlorure ferrique.
L'eau sortant du décanteur D est recueillie dans la cuve F pendant vingt-quatre heures; au bout de ce temps, on prélève un échantillon après avoir procédé à l'homogénéisation du mélange. La cuve F doit alors être nettoyée soigneusement.
1.6. Contrôle du dispositif de mesure.
La teneur en BiAS (en mg/l) de l'effluent synthétique est déterminée immédiatement avant usage.
La teneur en BiAS (en mg/l) de l'eau résiduaire collectée pendant vingt-quatre heures dans la cuve F doit être déterminée analytiquement par la même méthode, immédiatement après le prélèvement sinon les échantillons sont conservés, de préférence par congélation. La concentration doit être déterminée à 0,1 mg BiAS/l près.
Pour vérifier la bonne marche de l'opération, on mesure au moins deux fois par semaine la demande chimique en oxygène (DCO) ou le carbone organique dissous (COD) de l'effluent filtré sur fibre de verre, accumulé dans la cuve F et de l'effluent synthétique filtré qui est stocké dans la cuve A.
La diminution de la DCO ou du COD doit se stabiliser lorsque la biodégradation journalière du BiAS est à peu près régulière, c'est-à-dire à la fin de la période initiale indiquée sur la figure 3.
La teneur en matières sèches minérales de la boue activée contenue dans la cuve d'aération doit être déterminée deux fois par semaine en g/l. Si elle dépasse 2,5 g/l, il faut éliminer l'excès de boue activée.
L'essai de biodégradation est effectué à la température ambiante; cette température doit être régulière et maintenue entre 292 et 297 K (19-24° C).
1.7. Calcul de la biodégradation.
Le pourcentage de biodégradation de la BiAS doit être calculé quotidiennement à partir de la teneur en BiAS exprimée en mg/l de l'effluent synthétique et de l'eau résiduaire correspondante recueillie dans la cuve F.
Les valeurs ainsi obtenues doivent être représentées graphiquement, comme l'indique la figure 3.
La biodégradabilité du BiAS est calculée en prenant la moyenne arithmétique des valeurs obtenues au cours des vingt et un jours suivant la période initiale, délai pendant lequel la biodégradation doit avoir été régulière et l'installation doit avoir fonctionné sans aucune perturbation. En aucun cas, la durée de la période initiale ne dépassera six semaines.
Les valeurs quotidiennes de la biodégradation doivent être calculées à 0,1 % près, mais le résultat final est déterminé au nombre entier près.
Dans certains cas, la fréquence des prélèvements peut être diminuée mais, pour calculer la moyenne, on utilisera les résultats d'au moins 14 prélèvements journaliers répartis sur la période de vingt et un jours qui suivent la période initiale.
CHAPITRE 2. - Traitement préliminaire des produits à examiner
2.1. Notes préliminaires.
2.1.1. Traitement des échantillons
Le traitement des agents de surface non ioniques et des détergents préalablement à la détermination de la biodégradation par le test de confirmation est le suivant :
| Produits | Traitement |
| Agents de surface non ioniques Détergents |
Néant Extraction alcoolique suivie de la séparation des agents de surface non ioniques par échange d'ions |
Le but de l'extraction alcoolique est d'éliminer des produits commercialisés, les composants insolubles et inorganiques qui peuvent, le cas échéant, perturber le test de biodégradation.
2.1.2. Procédé d'échange d'ions.
Il est nécessaire, pour l'exactitude des tests de biodégradation, d'isoler et de séparer les agents de surface non ioniques du savon et des agents de surface anioniques et cationiques.
Ce résultat est obtenu grâce à l'application d'une technique d'échange d'ions utilisant une résine échangeuse macroporeuse et un agent d'élution approprié permettant l'élution fractionnée. Le savon et les agents de surface anioniques et non ioniques se trouvent ainsi isolés en une seule opération.
2.1.3. Contrôle analytique.
Après homogénéisation, on détermine la teneur en agents de surface anioniques et non ioniques du détergent suivant la méthode d'analyse à la MBAS et à la BiAS. La teneur en savon est déterminée selon une méthode appropriée.
Cette analyse du produit est nécessaire pour le calcul des quantités requises pour la préparation des fractions destinées aux essais de biodégradation.
Une extraction quantitative ne s'impose pas; toutefois, on extraira au moins 80 % des agents de surface non ioniques. Habituellement, on en obtient 90 % et plus.
2.2. Principe.
A partir d'un échantillon homogène (poudres, pâtes et liquides préalablement desséchés), on obtient un extrait par l'éthanol qui contient les agents de surface, le savon et d'autres composants solubles dans l'alcool, de l'échantillon de détergent.
L'extrait par l'éthanol est évaporé jusqu'à dessiccation complète et dissous dans un mélange isopropanol/eau; on fait passer la solution ainsi obtenue à travers un dispositif mixte échange de cations fortement acide/échange d'anions macroporeux, porté à la température de 323 K (50° C). Cette température élevée doit empêcher la précipitation des acides gras en milieu acide.
Les agents de surface non ioniques sont extraits de l'effluent par évaporation. Les agents de surface cationiques, susceptibles de perturber le test de biodégradation et la méthode analytique, sont éliminés par l'échangeur de cations placé au-dessus de l'échangeur d'anions.
2.3. Produits chimiques et appareillage.
2.3.1. Eau désionisée.
2.3.2. Ethanol, 95 % (v/v) C2H5OH (dénaturants admis : méthyléthylcétone ou méthanol).
2.3.3. Mélange isopropanol/eau (50/50 v/v).
50 parties d'isopropanol (CH3CHOH-CH3);
50 parties d'eau (2.3.1).
2.3.4. Solution de bicarbonate d'ammonium (60/40 v/v); 0,3 mol de NH4HCO3 dans 1000 ml d'un mélange isopropanol/eau constitué de 60 parties d'isopropanol et de 40 parties d'eau (2.3.1).
2.3.5. Echangeur de cation (KAT), fortement acide, résistant à l'alcool (50-100 mesh).
2.3.6. Echangeur d'anions (AAT), macroporeux, Merck Lewatit MP 7080 (70-150 mesh) ou équivalent.
2.3.7. Acide chlorhydrique, 10 % HCl (p/p).
2.3.8. Ballon à fond rond de 2 000 ml avec rodage conique et condenseur à reflux.
2.3.9. Entonnoir filtrant de 90 mm de diamètre (pouvant être chauffé) pour filtres en papier.
2.3.10. Fiole à vide de 2 000 ml.
2.3.11. Colonnes d'échangeurs à enveloppe chauffante et robinet; tube intérieur de 60 mm de diamètre et de 450 mm de hauteur (figure 4).
2.3.12. Bain-marie.
2.3.13. Etuve de séchage à vide.
2.3.14. Thermostat.
2.3.15. Evaporateur rotatif.
2.4. Préparation de l'extrait et séparation des agents non ioniques.
2.4.1. Préparation de l'extrait.
La quantité d'agents de surface nécessaire pour l'essai de biodégradation est d'environ 25 g BiAS.
Lors de la préparation des extraits pour les essais de biodégradation, la quantité de produit utilisée sera limitée à 2 000 g au maximum. De ce fait, il pourra être nécessaire de recommencer l'opération plusieurs fois afin d'obtenir une quantité suffisante pour l'essai de biodégradation.
L'expérience a montré qu'une série d'extractions limitées était préférable à l'extraction de grandes quantités.
2.4.2. Isolement des composants solubles dans l'alcool.
Ajouter 250 g du détergent à analyser à 1250 ml d'éthanol et porter le mélange à ébullition puis le soumettre à reflux pendant une heure, en agitant. Filtrer la solution alcoolique chaude dans un entonnoir filtrant à larges pores, porté à la température de 323 K (50° C) et aspirer fortement. Laver la fiole et l'entonnoir filtrant avec environ 200 ml d'éthanol chaud. Recueillir le filtrat et le lavage du filtre dans une fiole à vide.
Lorsque les produits à analyser sont des pâtes ou des liquides, s'assurer que l'échantillon ne contient pas plus de 25 g d'agents de surface anioniques et de 35 g de savon. Evaporer cet échantillon pesé jusqu'à dessiccation complète. Dissoudre le résidu dans 500 ml d'éthanol et procéder comme ci-dessus.
Dans le cas de poudres de faible densité apparente (< 300 g/l), il est recommandé d'augmenter la proportion d'éthanol dans le rapport 20/l.
Evaporer le filtrat d'éthanol jusqu'à dessiccation complète, de préférence au moyen d'un évaporateur rotatif. Répéter l'opération si une plus grande quantité d'extrait est nécessaire. Dissoudre la totalité des résidus dans 5 000 ml d'un mélange isopropanol/eau.
2.4.3. Préparation des colonnes d'échangeurs d'ions.
Colonne d'échangeurs de cations.
Placer 600 ml de résine échangeuse de cations (2.3.5) dans un bécher de 3 000 ml et couvrir en ajoutant 2 000 ml d'acide chlorhydrique (2.3.7). Laisser reposer pendant au moins deux heures en agitant de temps en temps. Décanter l'acide et transférer la résine dans la colonne (2.3.11) avec de l'eau désionisée.
La colonne doit comporter un tampon en laine de verre. Laver la colonne avec de l'eau désionisée à un débit de 10-30 ml/min. jusqu'à ce que l'éluat soit exempt de chlorure. Déplacer l'eau avec un mélange de 2 000 ml d'isopropanol/eau (2.3.3) à un débit de 10-30 ml/min. La colonne d'échangeurs est prête à l'emploi.
Colonne d'échangeurs d'anions.
Placer 600 ml de résine échangeuse d'anions (2.3.6) dans un bécher et l'immerger en totalité en ajoutant 2 000 ml d'eau désionissée. Laisser l'échangeur gonfler pendant au moins deux heures. Transférer la résine dans la colonne avec de l'eau désionisée. La colonne doit comporter un tampon en laine de verre.
Laver la colonne avec une solution 0,3 mol de monohydrogènocarbonate d'ammonium (2.3.4) jusqu'à ce qu'elle soit exempte de chlorure, ce qui nécessite environ 5 000 ml de solution. Laver ensuite avec 2 000 ml d'eau désionisée. Déplacer l'eau avec un mélange de 2 000 ml d'isopropanol/eau (2.3.3) à un débit de 10-30 ml/min. La colonne d'échangeurs est maintenant sous forme OH et prête à l'emploi.
2.4.4. Procédé d'échange d'ions.
Monter les colonnes d'échangeurs de sorte que la colonne d'échangeurs de cations se trouve au-dessus de la colonne d'échangeurs d'anions. Porter les colonnes à la température de 323 K (50° C) au moyen d'un thermostat. Chauffer 5 000 ml de la solution obtenue au point 2.4.2 jusqu'à 333 K (60° C) et passer la solution à travers le groupe d'échangeurs à un débit de 20 ml/min. Laver les colonnes avec un mélange chaud de 1000 ml d'isopropanol/eau (2.3.3).
Pour obtenir les agents de surface non ioniques, recueillir l'éluat et le lavage du filtre et les faire évaporer jusqu'à dessiccation complète, de préférence au moyen d'un évaporateur rotatif. Le résidu contient le BiAS. Ajouter de l'eau désionisée jusqu'à l'obtention d'un volume déterminé et doser la teneur en BiAS, conformément au point 3.3, dans l'aliquote. La solution est utilisée comme solution mère des agents de surface non ioniques pour l'essai de biodégradation. La solution doit être maintenue à une température inférieure à 278 K (5° C).
2.4.5. Régénération des résines échangeuses.
L'échangeur de cations est jeté après emploi.
On régénère la résine échangeuse d'anions en faisant passer dans la colonne environ 5000 - 6000 ml d'une solution de bicarbonate d'ammonium (2.3.4) à un débit d'environ 10 ml/min jusqu'à ce que l'éluat soit exempt d'agents de surface anioniques (essai au bleu de méthylène). Laver ensuite l'échangeur d'anions avec un mélange de 2 000 ml d'isopropanol/eau (2.3.3). L'échangeur d'anions peut de nouveau être utilisé.
CHAPITRE 3. - Dosage des agents de surface non ioniques dans les essais de biodégradation
3.1. Principe.
Les agents de surface sont concentrés et isolés par entraînement gazeux. Dans l'échantillon utilisé, la quantité d'agent de surface non ionique doit être de l'ordre de 250 - 800 µg.
L'agent de surface entraîné est dissous dans l'acétate d'éthyle.
Après séparation des phases et évaporation du solvant, l'agent de surface non ionique est précipité en solution aqueuse avec le réactif de Dragendorff modifié (KBil4 + BaCl2 + acide acétique glacial).
Le précipité est filtré, lavé avec de l'acide acétique glacial et dissous dans une solution de tartrate d'ammonium. Le bismuth présent dans la solution est dosé potentiométriquement avec une solution de pyrrolidinedithiocarbamate à pH 4-5, en utilisant une électrode indicatrice de platine poli et une électrode de référence au calomel ou d'argent, chlorure d'argent.
La méthode est applicable aux agents de surface non ioniques contenant 6-30 groupements d'oxyde d'alkène.
Le résultat du dosage est multiplié par le facteur empirique 54 de façon à exprimer arbitrairement les résultats en nonylphénol condensé avec 10 moles d'oxyde d'éthylène (NP 10).
3.2. Réactifs et appareillage.
Les réactifs doivent être préparés dans l'eau désionisée.
3.2.1. Acétate d'éthyle pur, fraîchement distillé.
3.2.2. Monohydrogénocarbonate de sodium NaHCO3 pour analyse.
3.2.3. Acide chlorhydrique (HCl) dilué (20 ml d'acide chlorhydrique pour analyse concentré dilué à 1000 ml avec de l'eau).
3.2.4. Méthanol pour analyse fraîchement distillé, conservé dans un flacon en verre.
3.2.5. Pourpre de bromocrésol (0,1 g dans 100 ml de méthanol).
3.2.6. Agent de précipitation : l'agent de précipitation est un mélange de 2 volumes de la solution A et 1 volume de la solution B. Le mélange est conservé dans un flacon en verre brun et peut être utilisé jusqu'à une semaine après sa préparation.
3.2.6.1. Solution A.
Dissoudre 1,7 g de nitrate basique de bismuth pour analyse (BiONO3-H2O) dans 20 ml d'acide acétique glacial et compléter avec de l'eau à 100 ml. Dissoudre ensuite 65 g d'iodure de potassium pour analyse dans 200 ml d'eau.
Mélanger ces deux solutions dans une fiole jaugée de 1000 ml, ajouter 200 ml d'acide acétique glacial (3.2.7) et compléter avec de l'eau à 1000 ml.
3.2.6.2. Solution B.
Dissoudre 290 g de chlorure de baryum (BaCl2-2H2O) pour analyse dans 1 000 ml d'eau.
3.2.7. Acide acétique glacial 99-100 % (des concentrations inférieures ne conviennent pas).
3.2.8. Solution de tartrate d'ammonium : mélanger 12,4 g d'acide tartrique pour analyse et 12,4 ml de solution aqueuse d'ammoniaque pour analyse (d =0,910 g/ml) et compléter à 1000 ml avec de l'eau (ou utiliser la quantité équivalente de tartrate d'ammonium pour analyse).
3.2.9. Diluer l'ammoniaque : diluer 40 ml d'ammoniaque pour analyse (d = 0,910 g/ml) avec de l'eau jusqu'à 1 000 ml.
3.2.10. Tampon acétate : dissoudre 40 g d'hydroxyde de sodium solide pour analyse dans 500 ml d'eau dans un bécher et refroidir. Ajoute : 120 ml d'acide acétique glacial (3.2.7). Bien mélanger, refroidir et transférer dans un ballon jaugé de 1 000 ml et ajuster au trait de jauge avec de l'eau.
3.2.11. Solution de pyrrolidinedithiocarbamate (ci-après dénommée "solution de carbate") : dissoudre 103 mg de pyrrolidinedithiocarbamate sodique (C5H3NNa S2-2H2O) dans environ 500 ml d'eau, ajouter 10 ml d'alcool n amylique pour analyse et 0,5 g de NaHCO3 pour analyse et compléter avec de l'eau à 1 000 ml.
3.2.12. Solution de sulfate de cuivre (pour étalonnage de 3.2.11).
Solution concentrée.
Dissoudre 1,249 g de sulfate de cuivre pour analyse (CuSO4-5H2O) avec 50 ml 0,5 mole d'acide sulfurique et compléter avec de l'eau à 1000 ml.
Solution étalon.
Mélanger 50 ml de solution concentrée avec 10 ml 0,5 mole H2SO4 et compléter avec de l'eau à 1 000 ml.
3.2.13. Chlorure de sodium pour analyse.
3.2.14. Appareil d'extraction des agents de surface (voir figure 5).
Le diamètre du disque fritté doit être identique au diamètre interne du cylindre.
3.2.15. Ampoule à décanter de 250 ml.
3.2.16. Agitateur magnétique avec aimant de 25-30 mm.
3.2.17. Creuset de Gooch, diamètre de la base perforée = 25 mm, type G 4.
3.2.18. Filtres circulaires en fibre de verre de 27 mm de diamètre; diamètre des fibres : 0,5 - 1,5 µm.
3.2.19. Deux fioles à vide avec allonges et collet de caoutchouc, de 500 ml et 250 ml respectivement.
3.2.20. Potentiomètre enregistreur équipé d'une électrode indicatrice de platine poli et d'une électrode de référence au calomel ou argent/chlorure d'argent permettant une gamme de mesure de 250 mV, et avec burette automatique d'une capacité de 20-25 ml ou dispositif manuel.
3.3. Méthode.
3.3.1. Concentration et séparation de l'agent de surface.
Filtrer l'échantillon aqueux à travers un papier filtre qualitatif. Eliminer les 100 premiers ml du filtrat.
Placer dans l'appareil d'extraction préalablement rincé à l'acétate d'éthyle une quantité mesurée de l'échantillon telle que celui-ci contienne entre 250 et 800 µg d'agent de surface non ionique.
Afin d'améliorer la séparation, ajouter 100 g de chlorure de sodium et 5 g de monohydrogénocarbonate de sodium.
Si le volume de l'échantillon dépasse 500 ml, ajouter ces sels sous forme solide dans l'appareil d'extraction et les dissoudre en faisant passer de l'azote ou de l'air dans l'appareil.
Si l'on utilise un échantillon de volume plus réduit, dissoudre les sels dans 400 ml d'eau puis les ajouter dans l'appareil d'extraction.
Ajouter de l'eau jusqu'à ce que le niveau atteigne le robinet supérieur.
Ajouter avec précaution 100 ml d'acétate d'éthyle à la surface de la phase aqueuse. Remplir le flacon laveur de l'arrivée de gaz (azote ou air) aux deux tiers d'acétate d'éthyle.
Faire passer dans l'appareil un débit de gaz de 30-60 l/h, l'emploi d'un rotamètre est recommandé. Le taux d'aération doit être progressivement augmenté au début. Le débit de gaz sera réglé de telle sorte que les phases restent bien séparées, de manière à limiter au minimum le mélange des phases et de la solution d'acétate d'éthyle dans l'eau. Couper l'arrivée de gaz après cinq minutes.
Si le volume de la phase organique diminue de plus de 20 % par dissolution dans l'eau, on répétera l'opération en diminuant le débit de gaz.
Verser la phase organique dans une ampoule à décanter. Reverser dans l'appareil d'extraction l'eau provenant de la phase aqueuse qui se trouverait dans l'ampoule à décanter (il ne devrait pas y en avoir plus de quelques ml). Filtrer la phase d'acétate d'éthyle à travers un papier filtre qualitatif sec dans un bécher de 250 ml.
Verser à nouveau 100 ml d'acétate d'éthyle dans l'appareil d'extraction et y faire passer de l'azote ou de l'air pendant cinq minutes. Soutirer la phase organique dans l'ampoule à décanter utilisée pour la première séparation, éliminer la phase aqueuse et faire passer la phase organique à travers le même filtre. Rincer l'ampoule à décanter et le filtre avec environ 20 ml d'acétate d'éthyle. Evaporer l'extrait d'acétate d'éthyle jusqu'à dessiccation complète sur un bain-marie (sorbonne). Diriger un léger courant d'air sur la surface de la solution pour accélérer l'évaporation.
3.3.2. Précipitation et filtration.
Dissoudre le résidu sec visé au point 3.3.1 dans 5 ml de méthanol, ajouter 40 ml d'eau et 0,5 ml de HCl dilué (3.2.3) et agiter le mélange avec un agitateur magnétique.
Ajouter à cette solution 30 ml de précipitant (3.2.6) avec une éprouvette graduée. Le précipité se forme par agitation. Après avoir agité pendant dix minutes, laisser le mélange reposer pendant au moins cinq minutes.
Filtrer le mélange dans un creuset de Gooch, dont la base est recouverte d'un filtre en fibre de verre. Puis laver le filtre sous faible dépression avec environ 2 ml d'acide acétique glacial. Ensuite, bien laver le bécher, le barreau aimanté et le creuset avec de l'acide acétique glacial (environ 40-50 ml). Il n'est pas nécessaire de transférer quantitativement sur le filtre le précipité qui adhère sur les parois du bécher parce que la solution du précipité destinée au titrage est reversée dans le bécher de précipitation, le précipité restant étant ensuite dissous.
3.3.3. Dissolution du précipité.
Dissoudre le précipité dans le creuset filtrant par addition à chaud (environ 353 K, 80° C) de la solution de tartrate d'ammonium (3.2.8) en trois fractions de 10 ml. Laisser chaque fraction reposer pendant quelques minutes dans le creuset avant de la filtrer dans la fiole.
Verser le contenu de la fiole filtrante dans le bécher utilisé pour la précipitation. Rincer les parois du bécher avec 20 ml de solution de tartrate pour dissoudre le reste du précipité.
Laver soigneusement le creuset, l'allonge et la fiole filtrante avec 150-200 ml d'eau et reverser l'eau de rinçage dans le bécher utilisé pour la précipitation.
3.3.4. Titrage.
Agiter la solution avec un agitateur magnétique (3.2.16), ajouter quelques gouttes de pourpre de bromocresol (3.2.5) et ajouter la solution d'ammoniaque diluée (3.2.9) jusqu'à obtention d'une coloration violette (la solution est légèrement acide compte tenu du résidu d'acide acétique utilisé pour le rinçage).
Ajouter ensuite 10 ml de tampon d'acétate (3.2.10), plonger les électrodes dans la solution et doser potentiométriquement avec la solution de carbate étalon (3.2.11), l'extrémité de la burette étant immergée dans la solution. La vitesse de titration ne doit pas dépasser 2 ml/min.
Le point d'équivalence est l'intersection des tangentes aux deux parties de la courbe du potentiel. On constatera à l'occasion que l'inflexion de la courbe du potentiel s'aplatit, ce à quoi on peut remédier en nettoyant soigneusement l'électrode de platine (par polissage au moyen d'un papier abrasif).
3.3.5. Témoin.
Simultanément, procéder à un dosage en blanc en suivant toute la méthode avec 5 ml de méthanol et 40 ml d'eau conformément aux instructions définies au point 3.3.2. Le dosage en blanc doit rester inférieur à 1 ml; sinon la pureté des réactifs 3.2.3. - 3.2.7 - 3.2.8 - 3.2.9 - 3.2.10) est suspecte (notamment leur teneur en métaux lourds) et il y a lieu de les remplacer. Il sera tenu compte du dosage en blanc dans le calcul des résultats.
3.3.6. Contrôle du facteur de la "solution de carbate".
Calculer chaque jour le facteur concernant la solution de carbate avant utilisation. A cet effet, doser 10 ml de la solution étalon de sulfate de cuivre (3.2.12) avec la solution de carbate après addition de 100 ml d'eau et 10 ml du tampon d'acétate (3.2.10). Si la quantité utilisée est égale à "a" ml, le facteur f s'obtient comme suite :
10
f = ----
a
et tous les résultats des dosages sont multipliés par ce facteur.
3.4. Calcul des résultats.
Chaque agent de surface non ionique a son propre facteur en fonction de sa composition, notamment de la longueur de la chaîne d'oxyde d'alkène. Les concentrations en agents de surface non ioniques sont exprimées par rapport à une substance de référence, un nonylphénol à 10 unités d'oxyde d'éthylène (NP 10) pour lequel le facteur de conversion est égal à 0,054.
La quantité d'agent de surface présente dans l'échantillon se trouve exprimée à l'aide de ce facteur comme suit :
0,054 f (b-c) = mg d'agent de surface non ionique exprimé en mg d'équivalent NP 10
où b = volume de solution de carbate utilisé pour l'échantillon (ml),
c = volume de solution de carbate utilisé pour le dosage en blanc (ml),
f = facteur de la solution de carbate.
3.5. Expression des résultats.
Exprimer les résultats en mg/l sous forme d'équivalent NP 10 à 0,1 mg près.
Figure 1
| A Récipient de stockage | E Pompe à air comprimé |
| B Pompe doseuse | F Collecteur |
| C Cuve d'aération (capacité 3 l) | G Aérateur (verre fritté) |
| D Décanteur | H Débitmètre à air |
Figure 2
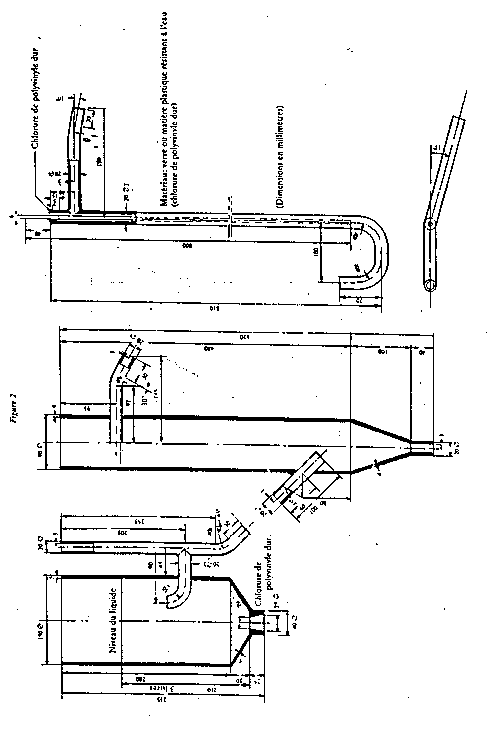
Figure 3
Calcul de la biodégradabilité - Test de confirmation
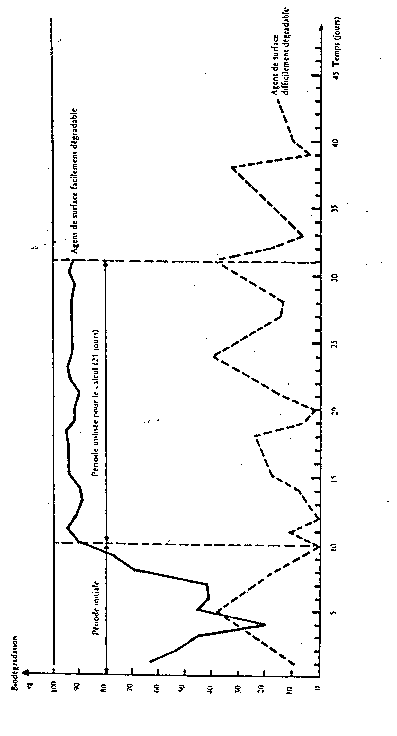
Figure 4
Colonne d'échangeur chauffante
(Dimensions en millimètres)
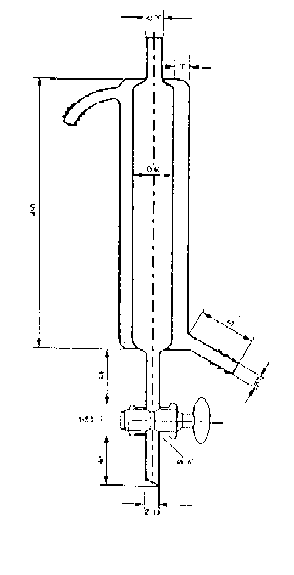
Figure 5
Vu pour être annexé à Notre arrêté du 25 octobre 1988.